




草铵膦,化学名称为2-氨基-4-[羟基(甲基)膦酰基]丁酸铵,是一种市场前景广阔的高效、低毒、广谱、非选择性的触杀型有机膦类除草剂,由德国赫斯特公司(拜耳)于20世纪80年代率先合成开发并于1986年上市。其纯品为结晶固体,具有微弱刺激性气味,不挥发、不降解、对光和空气稳定。
L-草铵膦(1)是消旋体草铵膦的活性异构体,而D-草铵膦(2)则基本无除草活性(图1)。L-草铵膦最初是从链霉菌中分离得到的一种单异构体的L-型氨基酸,该氨基酸是一种谷氨酰胺抑制剂,其除草活性是外消旋体草铵膦的2倍以上,也称为精草铵膦,因其分子结构与谷氨酸非常相似,故与谷氨酰胺合成酶的活性位点能发生可逆结合,有效抑制植物体内的L-谷氨酰胺合成,导致植物体内氮代谢紊乱,氨过量积累,叶绿体解体,从而使光合作用受抑制,最终导致植物死亡。L-草铵膦能够使草铵膦单位面积的用药量降低50%以上,在降低使用成本、减轻环境压力等方面成效显著。
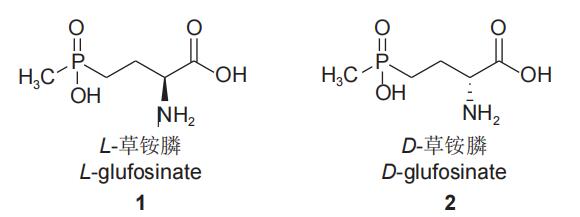
图1 L-草铵膦(1)和D-草铵膦(2)的结构式
目前,L-草铵膦的合成方法主要有化学合成法和生物催化法两大类,其中,化学合成法近年来得到较快发展,2016年,董文凯等根据当时已报道的专利及文献,基于手性中心不同的构建方法,对L-草铵膦的化学合成方法进行了综述;2020年,李嘉宁等根据已有的文献及专利,综述了合成草铵膦的不同工艺路线。本文通过对相关文献的调研、分析,以同一合成方法不重复综述为前提,总结了上述文献中未曾出现的L-草铵膦的化学合成方法。现有报道中,L-草铵膦的化学合成方法主要通过以下几种策略来实现:(1)L-脯氨酸衍生物辅助诱导合成;(2)以手性原料为手性源合成;(3)不对称催化合成。
1 L-脯氨酸衍生物辅助诱导合成
Soloshonok小组1992年报道了以L-脯氨酸为手性源诱导合成L-草铵膦的方法,L-脯氨酸先后与卤化苄和2-氨基二苯甲酮缩合,得到(S)-2-[N'-(N-苄基脯氨酰)氨基]二苯甲酮手性辅剂(3),随后把溶有甘氨酸的甲醇钠溶液滴加到溶有手性辅剂3和六水合硝酸镍的甲醇溶液中,50℃条件下反应2 h,以理想收率得到手性甘氨酸席夫碱NiII络合物(4),该络合物中甘氨酸的α-C在不同条件下可与β-氯乙基甲基膦酸酯和甲基乙烯基膦酸甲酯分别发生α-C的烷基化反应(图2)和Michael加成反应(图3),分别得到一对非对映异构体产物5和6。
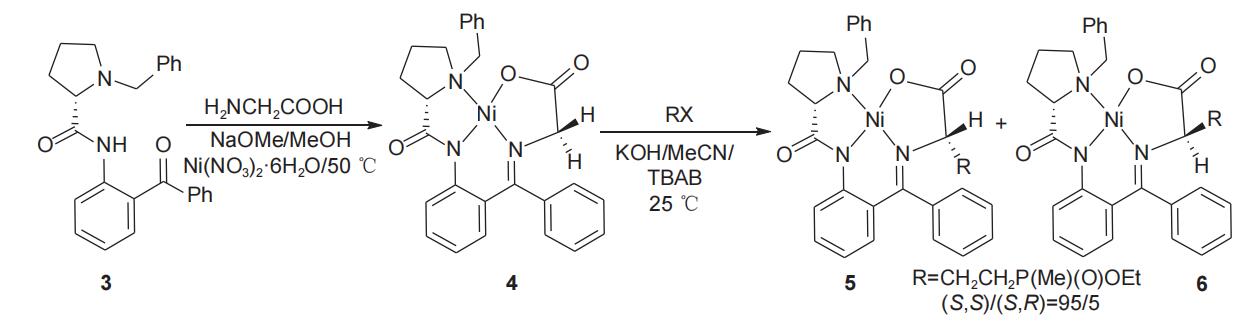
图2 L-脯氨酸衍生物诱导α-C的烷基化反应合成L-草铵膦前体
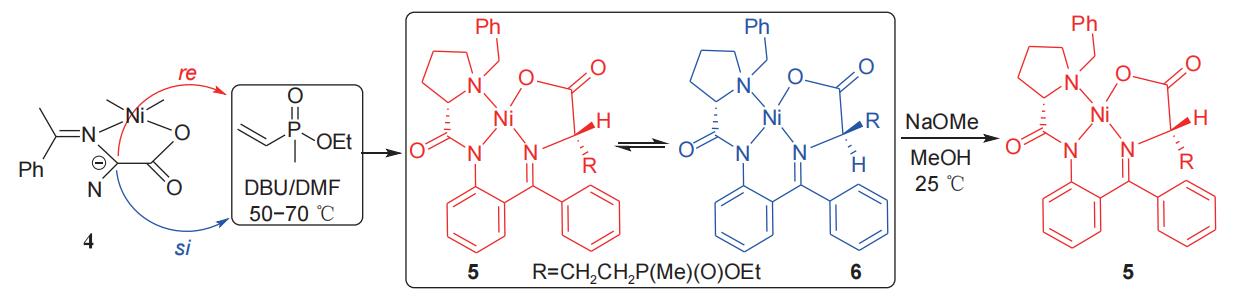
图3 L-脯氨酸衍生物诱导Michael加成反应合成L-草铵膦前体
在图2所示的反应式中,非对映异构体5/6的比例可达95/5,(S,S)-异构体(5)经酸解能以中等总收率(53%)和理想的比旋光度[α]58925=+16.8(c 0.5 g/L,H2O)得到L-草铵膦(1)并回收手性辅剂(S)-2-[N'-(N-苄基脯氨酰)氨基]二苯甲酮(3)(图4)。在图3中,非对映异构体5/6的比例为65/35,但将混合产物置于甲醇钠的甲醇溶液中,(S,R)-异构体(6)逐渐异构化为(S,S)-异构体(5),直到(S,R)-异构体(6)消失。该方法所得(S,S)-异构体(5)经酸解同样能够以中等总收率和理想的比旋光度得到L-草铵膦(1)并回收手性辅剂3。

图4 L-草铵膦前体酸解得到L-草铵膦
该法制备L-草铵膦(1)操作较为简单,反应条件较为温和,手性辅剂(3)经分离后可循环重复使用,但手性辅剂价格较高且反应需要使用(与手性辅剂)等物质的量的重金属镍盐,而镍盐若处理不当将对环境带来不利影响。
2 以手性原料为手性源合成
2.1 以L-丙氨酸为手性源
杨尚东研究小组2017年报道了钯催化惰性C(sp3)-H键烷基化高效合成手性γ-膦酰基-α-氨基酸的方法,该反应以L-丙氨酸为手性源,经与邻苯二甲酸酐和8-氨基喹啉作用,得到N-邻苯二甲酰保护的丙氨(8-氨基喹啉)酰胺关键中间体7,随后化合物7与甲基(碘甲基)膦酸乙酯在金属钯催化下偶联,得到L-草铵膦前体8,化合物8经脱保护、酸解等反应,高效合成了L-草铵膦(1)(图5)。马军安小组2021年对合成手性γ-膦酰基-α-氨基酸的方法从不同方面进行了较为全面的总结,为此类化合物的对映选择性合成提供了有益参考。
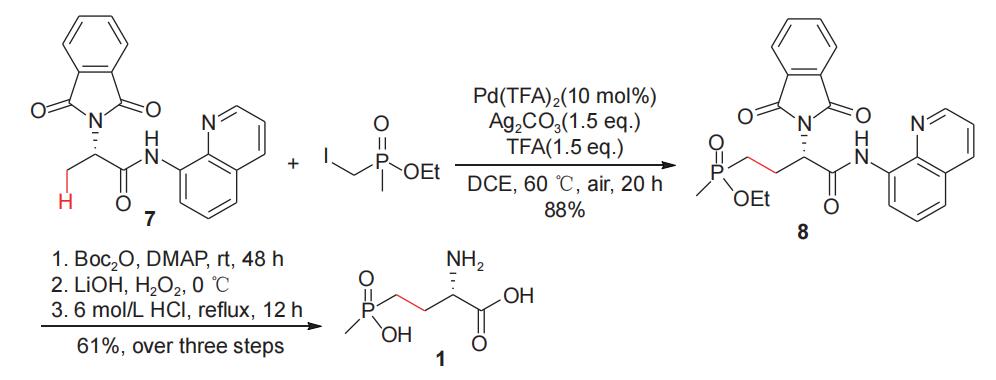
图5 L-丙氨酸诱导合成L-草铵膦
2.2 以L-谷氨酸为手性源
李超忠小组近期报道了以L-谷氨酸(9)为手性源,通过二碳酸二叔丁酯保护手性氨基,后经单甲酯化与N-羟基邻苯二甲酰亚胺在缩合剂和催化剂作用下进行缩合,使裸露羧基形成活化酯10,再与甲基亚膦酸二乙酯在光催化条件下进行脱羧膦酰化反应得L-草铵膦前体11,然后通过酸性水解、氨化反应得到目标产物L-草铵膦铵盐(图6)。产物的比旋光度为[α]D27=+16.2(c 1.00 g/L,H2O),纯度较理想。由于该合成路线步骤较多,致使总收率(30%)偏低。此外,该工艺中的封管过程具有一定的安全隐患,且需要价格昂贵的光催化剂,工业化前景较差。
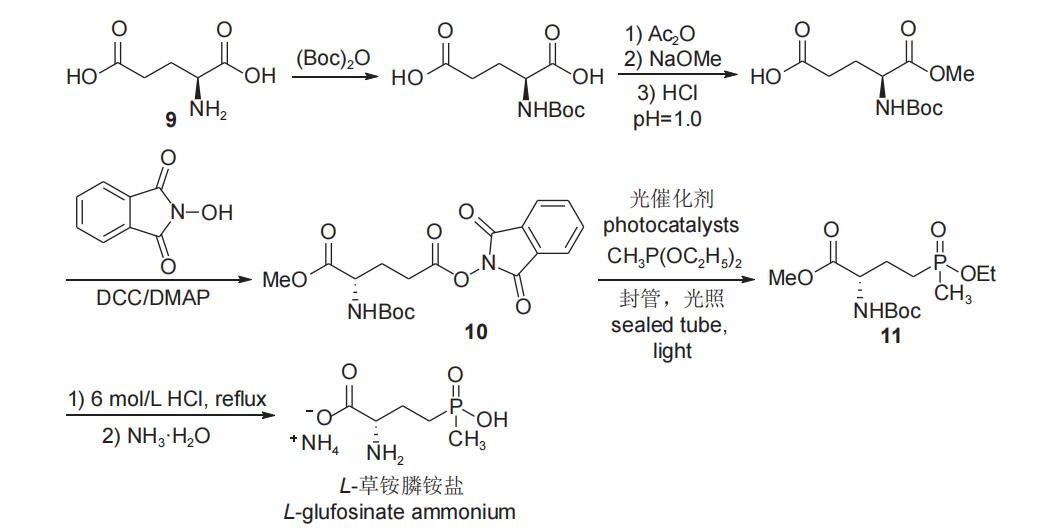
图6 以L-谷氨酸为手性原料合成L-草铵膦
2.3 以L-蛋氨酸为手性源
李旭坤等以L-蛋氨酸(12)为手性源,经与α-氯代羧酸或其衍生物反应得到L-高丝氨酸内酯盐酸盐(13),随后与氨基保护试剂氯甲酸酯反应得到氨基保护的L-高丝氨酸内酯(14),接着经开环得到β-氯乙基-L-甘氨酸衍生物15,化合物15与甲基亚膦酸二乙酯经Arbuzov反应,得到L-草铵膦衍生物16,水解后以69.2%总收率和93.5% e.e.值得到L-草铵膦(1)(图7)。该工艺较邱国福小组合成步骤有所简化,缩短了反应时间,提高了合成效率;另外,水相合成步骤有利于减轻环境负担,是一条较为有前景的合成工艺,但原料L-蛋氨酸价格偏高,同时会有含硫废物生成,增加了工艺成本。

图7 以L-蛋氨酸为手性原料合成L-草铵膦
2.4 以L-高丝氨酸为手性源
董文凯等2018年报道了以L-高丝氨酸(17)为手性源,经共沸脱水、氯代等步骤合成关键中间体L-3,6-双(β-氯乙基)-2,5-二酮哌嗪(18),随后18与甲基亚膦酸二酯经Arbuzov反应,得到L-草铵膦衍生物19,水解后以76.5%总收率和93.8%对映选择性得到L-草铵膦(1)(图8)。该合成路线操作较为简单易行,工艺三废较少,在脱水、卤代反应中分别使用酸性、亲核性催化剂,工艺过程较简单,提高了反应收率,工业化前景较好,但由于原料价格较高,不易控制生产成本。
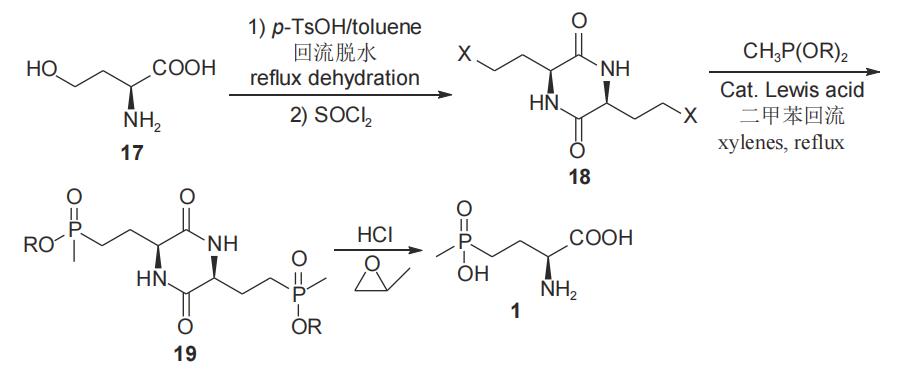
图8 以L-高丝氨酸为手性原料合成L-草铵膦
近两年,以L-高丝氨酸为手性源合成L-草铵膦的代表性工艺路线还有肖才根小组、王龙小组等的报道,采用相关工艺合成路线得到的L-草铵膦在总收率、对映选择性及产品纯度等方面均取得了一定的成功,具有一定的工业化应用价值,但由于原料价格较高,生产成本不易控制,在一定程度上限制了该工艺的发展。
2.5 以L-高丝氨酸内酯为手性源
1995年,Hoffmann报道了以L-高丝氨酸内酯(20)为手性源,经过开环氯代、酯化得到(S)-2-氨基-4-氯丁酸酯(21),化合物21与甲基亚磷酸二酯进行Arbuzov反应得到γ-膦酰基-α-氨基酸酯(22),随后经水解、精制,以中等总收率(43%)和较高的对映选择性(94.2% e.e.)得到L-草铵膦盐酸盐(图9),保护基不同对总收率略有影响。该工艺操作简便,但Arbuzov反应原料21的活性较低,需要在较高的温度(140℃)下才能进行,同时由于高温下氯代烷烃副产物进一步与甲基亚磷酸二酯发生副反应,使原料单耗增加。另外,高温反应还会使部分原料或产品消旋,导致L型e.e.值出现一定程度下降。为克服Arbuzov反应原料(21)活性较低的影响,刘永江等于2021年报道了以L-高丝氨酸内酯盐酸盐为手性源,经开环、酯化得到化合物21,随后由氯(乙氧基)(甲基)膦(该化合物在反应体系中先由甲基二氯化膦与甲基亚膦酸二乙酯反应生成)代替甲基亚磷酸二乙酯与化合物21反应,温度(100℃)明显降低,且L-草铵膦的总收率(71%)和对映选择性(98% e.e.)均有提高(图10)。该工艺路线前景较好,但由于原料价格较高,产品成本不易控制。
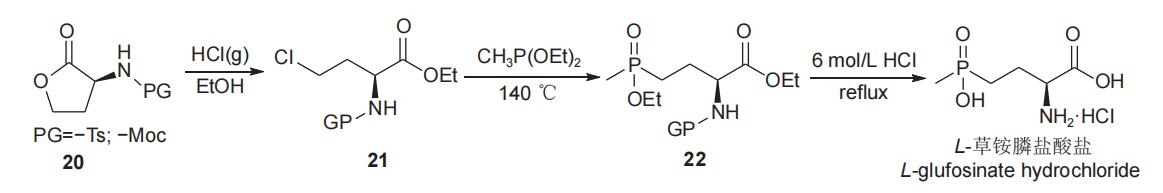
图9 以L-高丝氨酸内酯为手性原料合成L-草铵膦
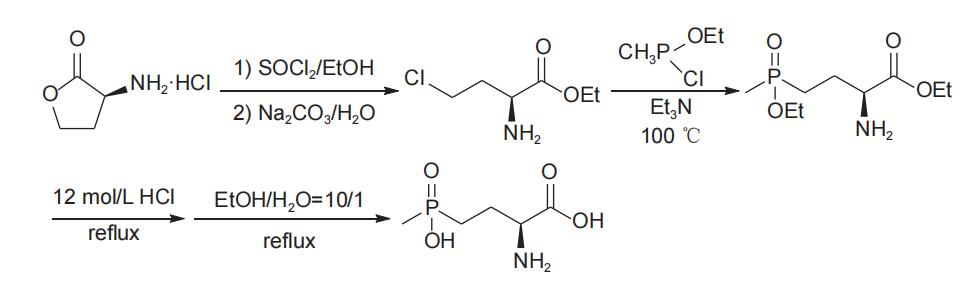
图10 以L-高丝氨酸内酯盐酸盐为手性原料合成L-草铵膦
近期,汤文杰等对含磷反应物进行了系统优化,获得了系列高活性含磷底物,如:甲基亚磷酰二胺、甲基亚磷酰胺单酯、氯(甲基)亚膦酰胺等,与(S)-2-氨基-4-氯丁酸酯(21)可在90℃左右完成反应,且均能以较高总收率和理想的对映选择性获得L-草铵膦。上述更高反应活性底物的开发,使得P-C键的构建条件更为温和,降低了反应温度,可减少L-构型在高温下的消旋,从而提高了L-草铵膦的纯度。相应地,也省去了Arbuzov反应过程中所产生副产品的分离、纯化和收集等环节的投入,具有较好的工业化前景,但由于原料价格较高,不易控制产品成本,一定程度上限制了该工艺的发展。
2.6 以L-乙烯基甘氨酸为手性源
2020年,程柯等报道了以L-3,6-双乙烯基-2,5-二酮哌嗪(22)为起始物,在引发剂过氧化苯甲酸叔丁酯存在下与甲基次膦酸酯反应获得L-草铵膦前体23,化合物23直接水解即可以高达90%的收率、97% e.e.值得到L-草铵膦(1)。该反应起始物(22)为L-乙烯基甘氨酸(24)的二聚体,因其溶解度较差,反应体系温度需要控制在90℃以上。为解决上述问题,2022年李南等报道了以L-乙烯基甘氨酸或其衍生物24在引发剂过氧化新戊酸叔丁酯存在下与甲基次膦酸酯于75℃条件下反应获得中间体25,25直接水解即可以高达93%的收率、98% e.e.值得到L-草铵膦(1)(图11)。上述两种工业化生产L-草铵膦方法的收率和对映选择性均非常理想,但由于24不是天然氨基酸,需要由价格较高的L-蛋氨酸或L-高丝氨酸合成得到,生产成本偏高。
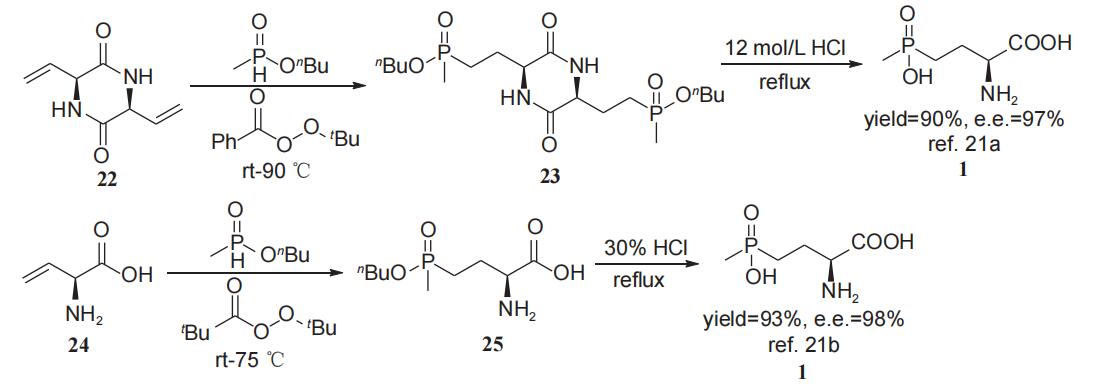
图11 以L-乙烯基甘氨酸为手性原料合成L-草铵膦
3 不对称催化生成
闫立单等在2016年报道了以4-(羟基-(甲基)氧膦基)-2-乙酰氧基丁腈(26)为起始原料,经过氰基水解、α-羟基氧化、羰基亚氨化过程分别获得化合物27~29,化合物29通过L-型手性催化剂—三苯基膦-氯化铑不对称催化加氢合成L-草铵膦(1),该工艺路线较为简单,以超过80%的收率和92%以上的对映选择性得到L-草铵膦(1)(图12)。与目前国外工业化生产工艺相比,该工艺所得产品的收率及对映选择性均有一定提高,但该工艺起始原料不易得,同时反应经历了金属钌催化氧化、高压手性铑催化加氢等步骤,一定程度上限制了该工艺的应用。
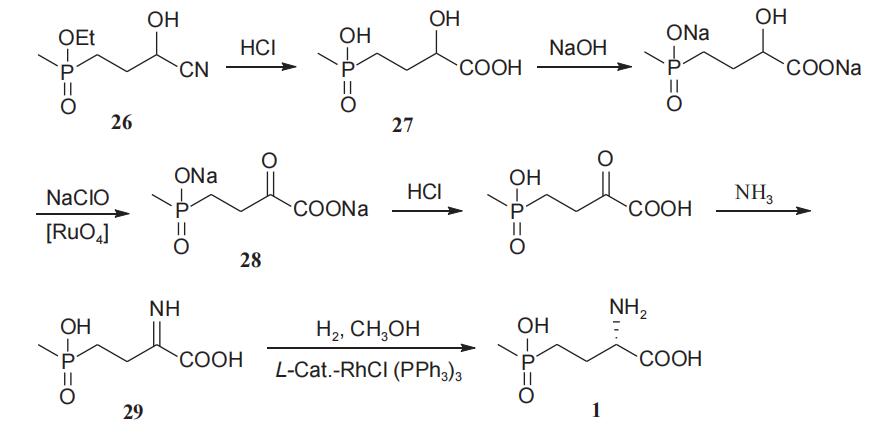
图12 不对称催化氢化法制备L-草铵膦
4 总结与展望
本文以总结L-草铵膦的化学合成方法为目标,通过对相关文献的进行分析、归纳,就L-脯氨酸衍生物辅助诱导合成、以手性原料为手性源合成以及不对称催化合成等合成方法,分类介绍了L-草铵膦的合成工艺及优缺点,为今后化学方法合成L-草铵膦提供参考。
草铵膦作为全球第二大转基因作物除草剂,具有杀草谱广、低毒、活性高和环境相容性好等特点,其发挥活性作用的速度比百草枯慢而优于草甘膦。在百草枯禁用和草甘膦抗性问题日益凸显的背景下,草铵膦应用前景广阔。而L-草铵膦相较于传统草铵膦,其活性倍增、用量减半,正契合了国家农药减量增效政策。目前,L-草铵膦的不对称催化加氢合成工艺已在日本实现工业化生产,但在中国由于其生产成本较高,尚缺乏市场竞争力。因此,开发反应条件温和、收率高、对映选择性好且成本低的L-草铵膦合成工艺仍是科技工作者今后努力的方向。
(1)本网旨在传播信息,促进交流,多方面了解农药发展动态,但不构成任何投资建议。
(2)所有文章仅代表作者观点,不代表本网立场。
(3)“信息来源:江苏省农药协会 农药资讯网”为原创文章,转载时请注明来源和作者。
(4)本网转载文章及图片的版权属于原作者,若有侵权,请联系删除。