




琥珀酸脱氢酶抑制剂(SDHI)类杀菌剂在防治谷物病害方面具有重要的作用,可有效防治小麦叶枯病、小麦叶锈病或大麦云纹病等大多数谷物重要病害。
Isoflucypram(开发代号BCS-CN88460,商品名iblon)是拜耳作物科学公司开发的新型琥珀酸脱氢酶抑制剂(SDHI)类杀菌剂,其持效期长,可延长谷物灌浆时间。Isoflucypram于2019在新西兰首获登记,用于防治大麦网斑病、叶斑病、叶锈病,小麦条锈病、叶锈病、叶枯病,黑麦条锈病和叶锈病等,可提高谷物产量和品质。
1 理化性质
Isoflucypram的IUPAC化学名称:N-(5-氯-2-异丙基苄基)-N-环丙基-3-(二氟甲基)-5-氟-1-甲基-1H-吡唑-4-甲酰胺;CAS化学名称:N-[[5-氯-2-(1-甲基乙基)苯基]甲基]-N-环丙基-3-(二氟甲基)-5-氟-1-甲基-1H-吡唑-4-甲酰胺。CAS登录号:1255734-28-1。

图1 Isoflucypram结构式
在20℃和101.3 kPa,纯品为无味白色粉末,原药为具有轻微气味的浅米色粉末。熔点108.8℃,在215℃开始分解;相对密度:纯品D420=1.22,原药D420=1.31;蒸气压:1.2×10-7 Pa(20℃)、2.8×10-7 Pa(25℃)、1.5×10-5 Pa(50℃);表面张力σ=68.2 mN/m(90%饱和溶液,20℃)。水中溶解度:1.8 mg/L(20℃,蒸馏水,pH 5.8);有机溶剂中的溶解度(20℃,g/L):正庚烷1.2,甲苯>>260,二氯甲烷>260,甲醇97,丙酮>260,乙酸乙酯>260,二甲亚砜>260。分配系数:logPow=4.0(25℃)。
2 毒性
原药(94.2%)对大鼠急性经口LD50>2,000 mg/kg,对大鼠急性经皮LD50>2,000 mg/kg,对雄性大鼠急性吸入LC50为3.131 mg/L,对雌性大鼠急性吸入LC50为2.209 mg/L。对兔皮肤无刺激性,对兔眼睛没有刺激作用,会引起小鼠皮肤过敏反应。对哺乳动物没有基因毒性、致癌性,对发育、繁殖没有影响,没有神经毒性。对黑头呆鱼(Pimephales Promelas)的LC50(96 h)为0.0861 mg/L,虹鳟LC50(96 h)为0.104 mg/L,对大型溞EC50(48 h)0.201 mg/L,羊角月牙藻EC50(72 h)>2.02 mg/L,浮萍EC50(7d)>2.48 mg/L。Isoflucypram对水生生物毒性高,而且作用时间长。
对蜜蜂的毒性:触杀毒性LD50(48 h)>100.0 μg a.i./只,经口毒性LD50(48 h)>106.3 μg a.i./只;对山齿鹑急性经口LD50>2,000 mg a.i./kg bw,对绿头鸭的急性经皮LD50>1,360 mg a.i./kg bw;对安德爱胜蚓LC50>1,000 mg a.i./kg bw(土壤)。
由上述内容可知,isoflucypram对哺乳动物具有较低的急性毒性,对水生生物毒性很高,对陆生生物没有毒性。
Isoflucypram在水生环境和土壤环境中持效期长,在土壤中的迁移能力低,生物积累性弱。
3 创制过程
Isoflucypram的发现可追溯到通用结构为A的磺酰甲酰胺(图2),此类物质是经化学库设计合成,是早期先导物,其活性谱窄,对白粉病、褐锈病、网斑病等真菌病害有效。为了发现新的除草剂或杀虫剂,Desbordes Philippe等人把Rohn & Haas发现的除草活性结构A1和杀虫剂氟啶虫酰胺(A2)拼接,同时对A1中的炔丙胺基或A3中的氰甲胺基进行优化,如替换为烯丙胺、异丙胺和环丙胺等小的胺基团。取代为环丙胺后发现了氟喹啉类抗生素环丙沙星(A4)。把A3结构中的4-(三氟甲基)吡啶-3-甲酰胺部分衍生为五元杂环,特别是吡唑杂环,众所周知吡唑是是氟唑菌苯胺(A5)等杀菌剂的核心部分。

图2 杀菌剂磺酰甲酰胺化学结构的发现
A6是被发现的第一个具有温室盆栽杀菌生物活性的磺酰甲酰胺化合物,其作用机制未知。以A6为先导,对芳基磺酸、氨基和吡唑酸进行衍生优化(图3)。结果发现3-取代吡唑甲酸(A7a-c),特别是5-氟吡唑-4-基甲酸(A7a)对杀菌活性重要,其直接作用于SDH。环丙胺基是维持活性的必要因素,而其他氨基取代物活性降低或活性消失。对亲脂性磺酸部分的修饰发现邻位氯代苯基(A8)和对位苯氧取代的苯基(A9)化合物活性增加。
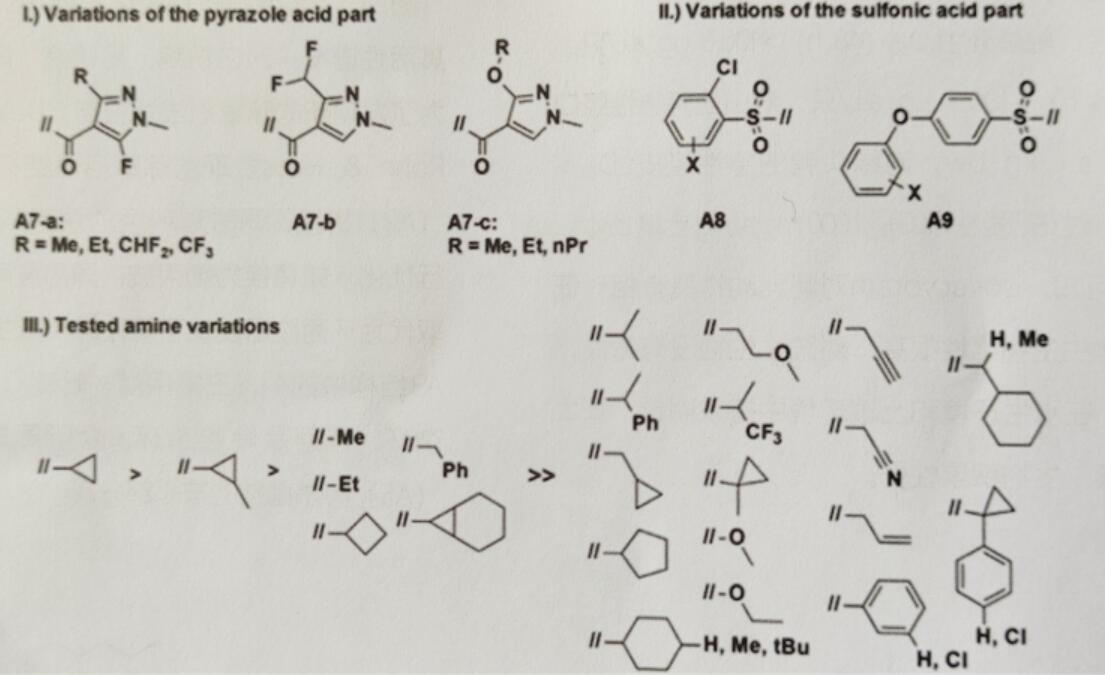
图3 具有杀菌活性的磺酰甲酰胺:从苗头(hit)化合物到先导物的修饰过程
把含氯亲脂磺酸基(A8)和3-取代吡唑甲酸基(A7a-c)合并得到了在温室生测中杀菌活性提高的化合物A10和A11(图4)。对A11进行了田间谷物病害生物活性测定,试验结果为即使高的使用剂量,化合物的活性也弱。
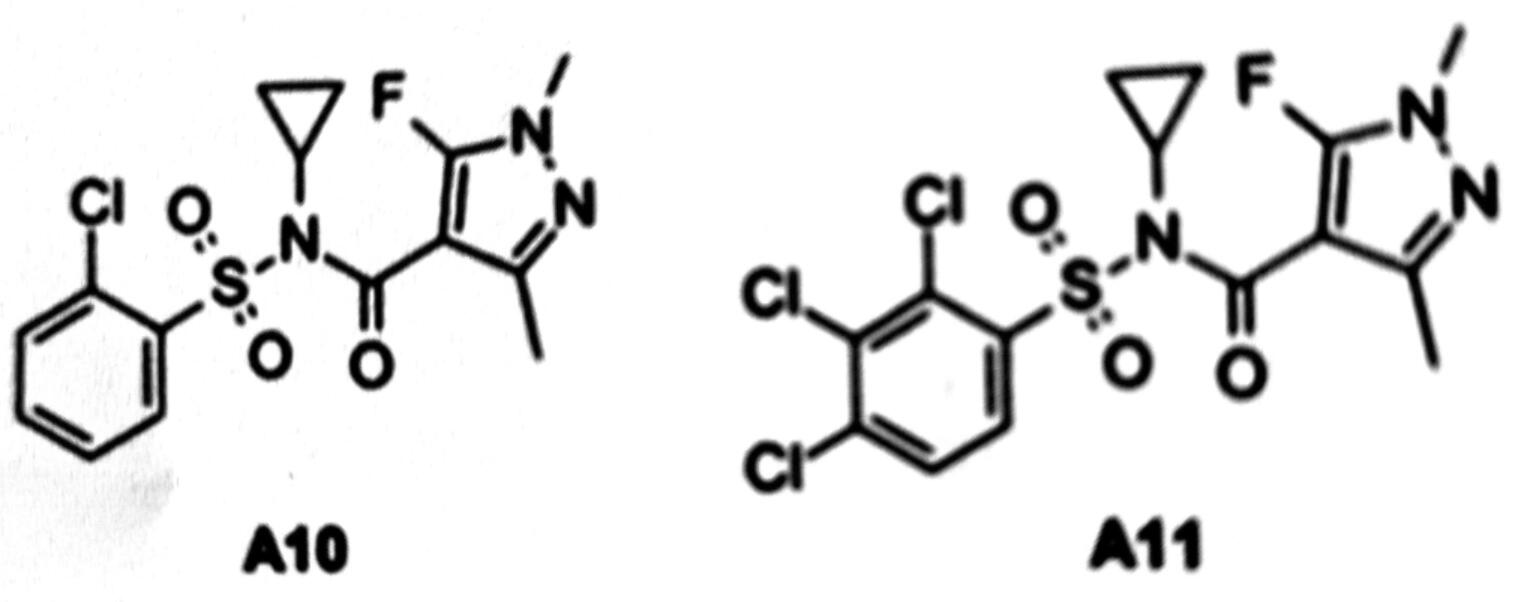
图4 具有杀菌活性的磺酰甲酰胺:先导物结构
A10对黑粉菌属SDH的离体活性中等,但对葡萄孢属、Zymoseptoria和伞菌属(Agaricus)菌的SDH有强的抑制作用。强的离体活性表明环丙基取代物不是药物前体,此取代基是A10与受体结合的必要组成部分。然而,发现A10和A11在植物体内很快降解,活性降低。在此发现的基础上,对磺酰甲酰胺进行更多的修饰研究。
为了解决化合物A11在田间活性差的问题,对磺酰胺基团进行了进一步的修饰。羰基或硫原子取代磺酰基团产生的化合物无活性,而烷基取代对离体和温室生测杀菌活性有大的影响。把氟吡菌胺A12的亲脂侧链结合到化合物A11的N-环丙基酰胺基团上得到了新化学类别化合物A13,此化合物对数种真菌病原菌的SDH有很强的抑制活性(图5)。很快研究发现化合物A13的吡啶环能被苯环取代,且必须要很好地控
制苄基基团和N-环丙基间的二面角。这可通过邻位取代大的基团和/或苄基来实现。而在邻位的再取代会增加此效应,由此取代产生化合物A14和A15。
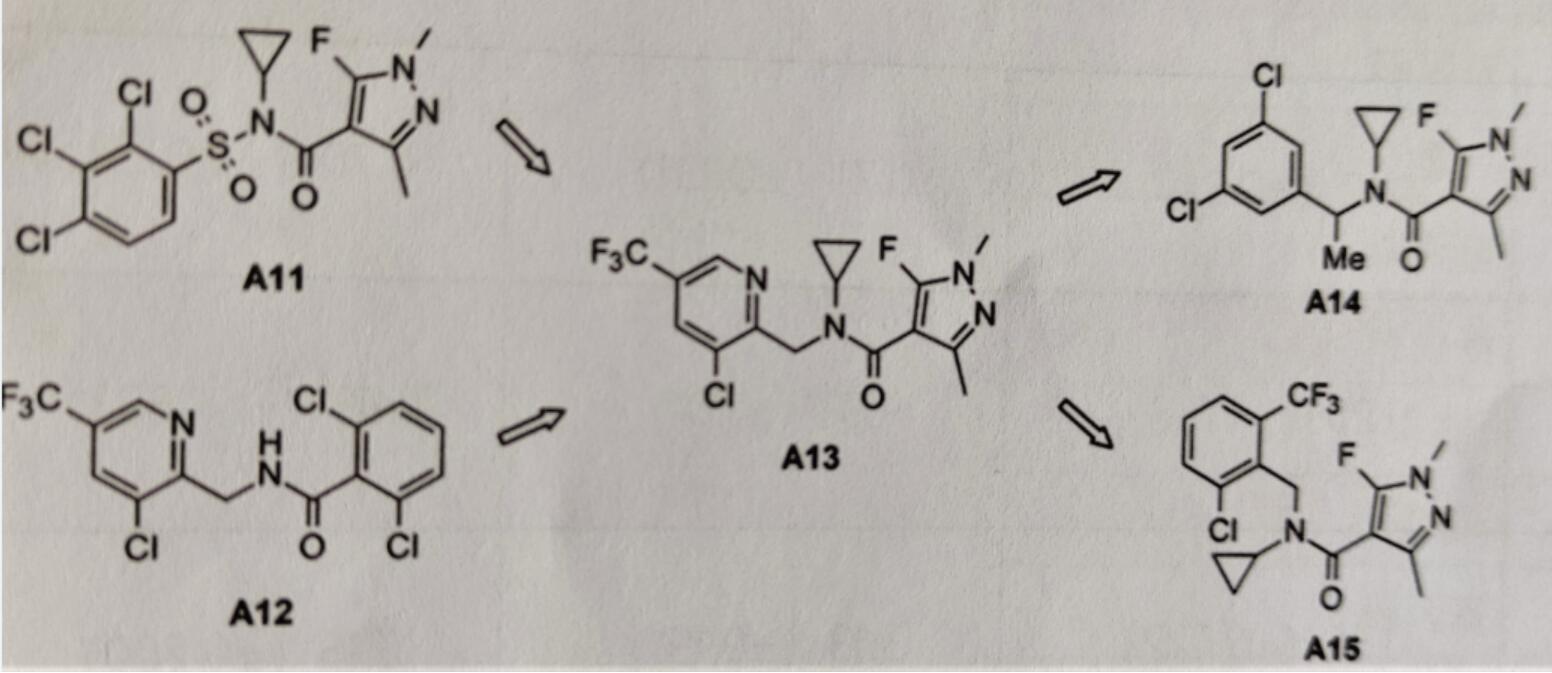
图5 具有杀菌活性的N-苄基-N-环丙基甲酰胺和其首次优化
对酸部分进行了充分的研究,以200多个5元或6元环取代最初的5-氟-1,3-二甲基-1H-吡唑-4-甲酸(A7a,R=CH3)。发现已知SDHI抑制剂具有的典型吡唑结构为A7a(R=CH3)和A7b,使化合物具有广谱活性。此外,注意到吡唑环的-5-氟取代有重要作用,故评估了含有由效果最好两个酸——A7a(R=CH3)和A7b结合所得的酸A7a(R=CHF2)的化合物的活性(图6)。此酸部分使化合物对SDH的抑制活性提高了1~2个数量级,活体试验的ED50降低了。
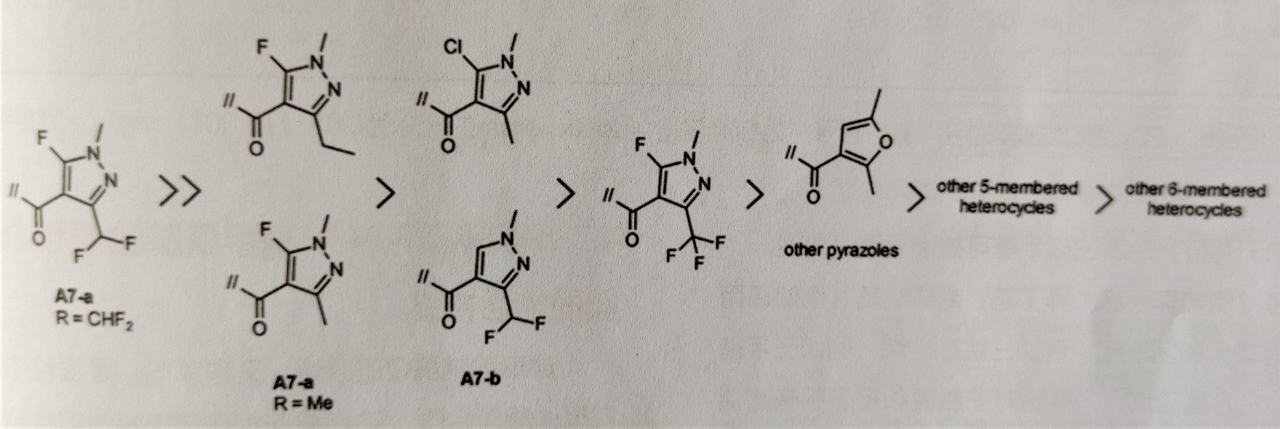
图6 酸部分的构效关系
对于磺酰甲酰胺,2-甲基环丙基取代基除了是活性部分外,环丙基几乎是酰胺氮仅能接纳的取代基。这再次证实N-环丙基衍生物不是药物前体,因为发现几乎所有NH类似物无活性。相反,发现硫代酰胺是酰胺的真正药物前体,硫代酰胺在植物体内具有相似水平的活性,但直接作用于靶标没有活性。
化合物A15(图7)的2-三氟甲基可被卤元素(如溴、碘),小烷基(例如异丙基、异丁基)或环烷基(例如环丙基),甚至是较大基团取代,例如三烷基硅烷(三甲基硅烷[TMS]、三乙基硅烷),得到对子囊菌和担子菌有高活性的化合物。邻苯取代物的构效关系(SAR)为2-iPr>2-CF3≥2-I>2-TMS。A16的2-异丙基取代衍生物对Zymoseptoria tritici细胞的ED50最小(表1),且对细胞的活性和温室、大田试验活性相差无几。
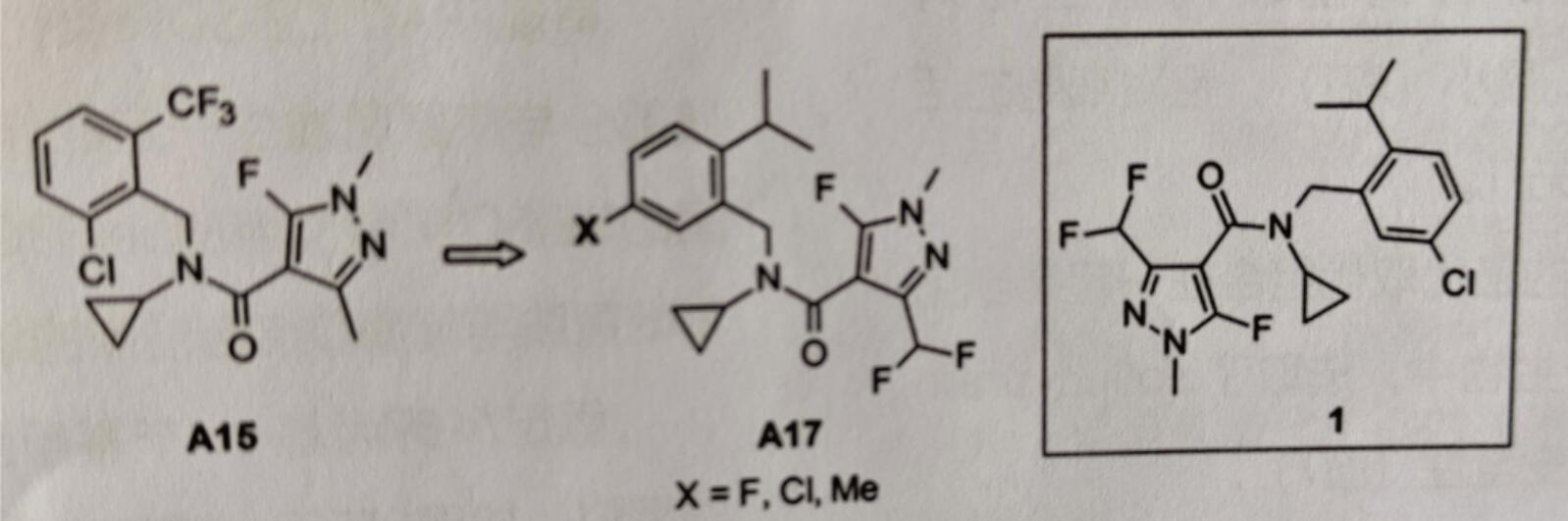
图7 对具有杀菌活性的N-苄基-N-环丙基甲酰胺的进一步优化得到isoflucypram(1)
表1 A16衍生物对Zymoseptoria tritici
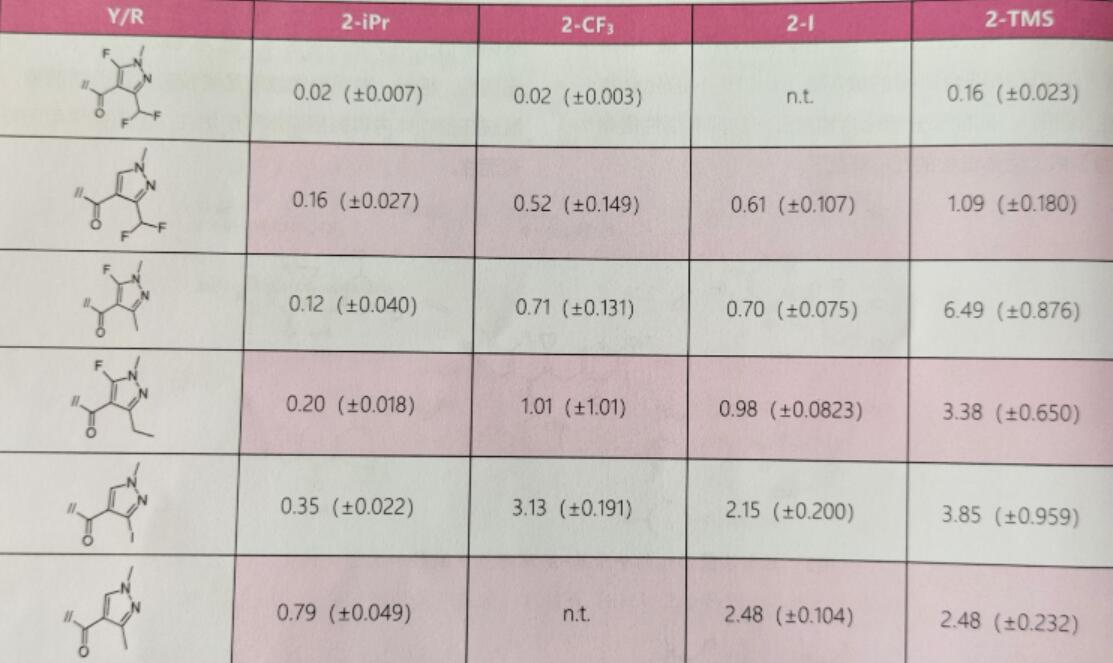
注:n.t,没测试;ED50为8次重复的平均值;括号中为标准偏差;isoflucypram的ED50为(0.05±0.008)mg/L,为参考。
对苯基、酰胺和吡唑基团的最优取代组合在一起得到化合物A17。在这些高活性的衍生物中,选取了isoflucypram(1),此物质具有较好的活性和安全性(图7)。
4 N-环丙基-N-苄基甲酰胺的亲和力数据和可能的结合方式
琥珀酸辅酶Q还原酶(又称SDH)是线粒体内膜上的异四聚体酶复合体。SDHI杀菌剂作用于病原菌线粒体内膜上由亚基B、C、D组成的辅酶Q。
再观FRAC C2-SDHI类化合物,大多数SDHI通过其甲酰胺部分与病原菌发生一定的作用。据推测,甲酰胺氢与病原菌的辅酶Q结合位点的Ser83C形成水介导的氢键。图8为联苯吡菌胺与辅酶Q结合位点的结合方式。
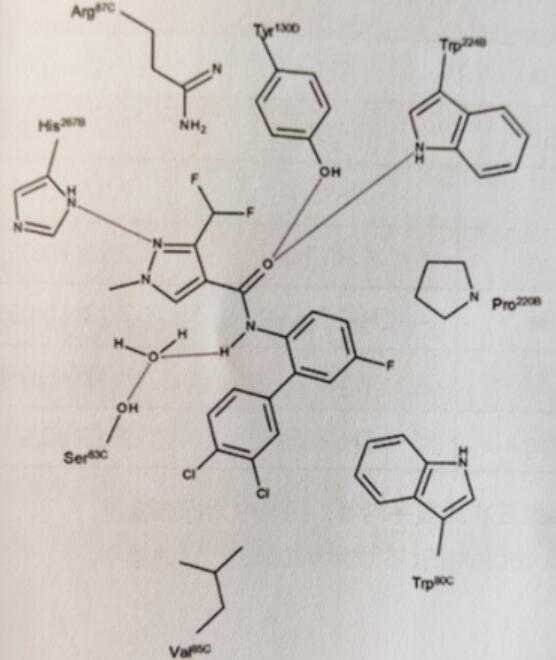
图8 推测联苯吡菌胺与Zymoseptoria tritici辅酶Q结合位点的二维结合方式
假定环丙基N-苄基甲酰胺的结合方式与典型的SDHI的相似,如图9所示。甲酰胺氧原子与保守的色氨酸NH侧链形成氢键,吡唑基团与组氨酸NH侧链形成另一氢键。氯取代苯环可能结合到真菌和线虫特有的疏水口袋中。

图9 推测isoflucypram与Zymoseptoria tritici辅酶Q结合位点的二维结合方式
然而,与典型的SDHI相比,N-环丙基不支持水介导的氢结合到Ser83C。在通用结构2的基础上合成了含有不同N-取代基(X)的数个化合物,并测定了这些物质对灰葡萄孢菌(Botrytis cinerea)的复合体酶的抑制活性(表2)。
表2 通用结构2衍生物对灰葡萄孢菌复合体Ⅱ酶的pl50
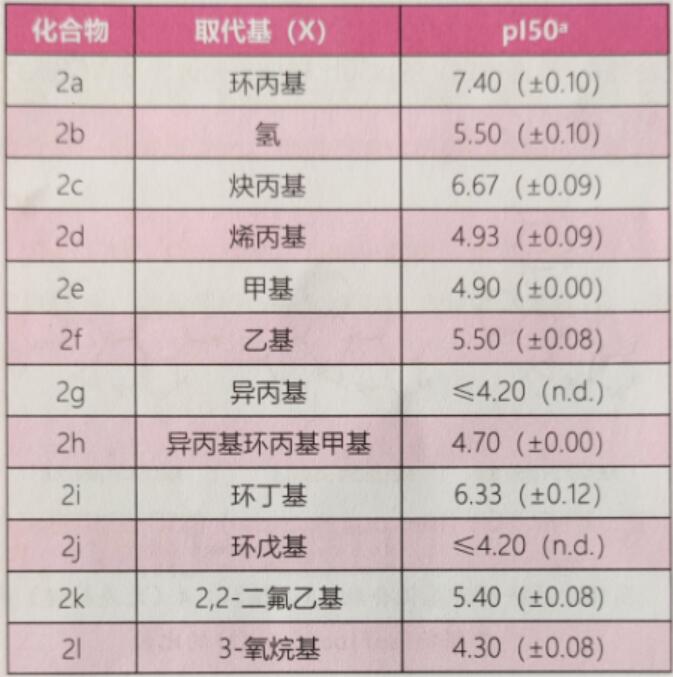
注:a为3次重复平均值;括号中为标准偏差;isoflucypram的pl50为(8.6±0.0),为参考。
如果N-环丙基-N-苄基甲酰胺各方面的表现都和常见的SDHI一样,预期NH衍生物2b活性要高于N-环丙基衍生物2a。但是,虽然2a不能形成上述所提及的水介导的氢键,它也具有很大的pl50,显著高于2b的抑制活性。化合物的活性因N-取代基不同而不同。当环从N-环丙基(2a)变为N-环丁基(2i),即环的大小增加时,pl50降低一个数量级;变为N-环戊基(2j),pl50降低3个数量级。把环丙基环打开为N-异丙基(2g),所得化合物几乎没有离体活性。总的来说,N-甲酰胺取代的构效关系非常清晰,N-环丙基化合物的活性最好。
pl50增加可能是由于在结合口袋中失去1个水分子所致。熵增益远远弥补了水介导氢与Ser83C结合的缺乏,如化合物1具有极大的pl50所表明的。
除环丙基基团外,N-环丙基-N-苄基甲酰胺具有前所未有的次级结构模式——苯基和甲酰胺基团间的C1链。
所有已知的SDHI要么不含有连接链(如啶酰菌胺、联苯吡菌胺或氟唑菌酰胺的吡唑-4-甲酰胺),要么不含有C2链(氟吡菌酰胺和氟唑菌酰羟胺)。对化合物3(C1链)和化合物4(无连接链)的pl50比较后优先选择具有C1链的化合物3(图10)。

注:对Neurospora crassa的SDH测定的pl50。
图10 对N-环丙基化合物3(C1链)、4(无连接链)和参照物isoflucypram(1)的比较
与N-环丙基的清晰的离体构效关系相比,结构5的化合物链的延伸(A)没有明显的构效关系(表3)。
所有(未)取代的C1和C2链与N-环丙基组合的化合物都具有高的离体活性。含有未取代C1链的化合物具有较高的温室和田间活性以及广谱杀菌活性。
对SDH抑制剂以前所未有的方式进行取代得到了C2-琥珀酸脱氢酶抑制剂类别的一个新子类杀菌剂isoflucypram,也称为N-环丙基-N-苄基甲酰胺。与其他SDHI相比,isoflucypram结合方式的改变是否使其与其他SDHI不具有交互抗性,这还需要进行进一步的研究。
表3 基于通用结构5的化合物对Botrytis cinerea复合体酶Ⅱ的pl50

注:a为3次重复平均值;括号中为标准偏差;isoflucypram的p150为(8.6±0.0),为参考。
5 总结
Isoflucypram广谱,对哺乳动物低毒,可能是谷物杀菌剂市场新的标杆——能很好地、持效地防治所有相关的叶部病害,增加作物产量。Isoflucypram是在丰富的SDHI化学知识的基础上对不同结构基团进行结合,化学优化得到的物质。Isoflucypram的生化构效关系表明其与SDH辅酶Q结合。2个独特的结构基团——N-环丙基、甲酰胺和苯基间的C1链使isoflucypram成为FRAC复合体Ⅱ抑制剂类的子类。
(1)本网旨在传播信息,促进交流,多方面了解农药发展动态,但不构成任何投资建议。
(2)所有文章仅代表作者观点,不代表本网立场。
(3)“信息来源:江苏省农药协会 农药资讯网”为原创文章,转载时请注明来源和作者。
(4)本网转载文章及图片的版权属于原作者,若有侵权,请联系删除。